Result Interpretation#
Output Files#
The mDeepFRI predict-function command generates several output files:
Alignment Summary#
File: alignment_summary.tsv
Summary statistics of sequence-structure alignments:
Query sequences: Number of query proteins processed
Aligned sequences: Number of proteins with successful alignments
Alignment rate: Percentage of successful alignments
Average identity: Mean sequence identity across all alignments
Average coverage: Mean query coverage across all alignments
Database Search Results#
Directory: database_search/
Raw MMseqs2 search results for each query protein.
Prediction Matrices (optional)#
Files: prediction_matrix_bp.tsv, prediction_matrix_cc.tsv, prediction_matrix_mf.tsv, prediction_matrix_ec.tsv
Detailed prediction matrices for each ontology mode:
prediction_matrix_bp.tsv- Biological Process predictionsprediction_matrix_cc.tsv- Cellular Component predictionsprediction_matrix_ec.tsv- Enzyme Commission predictionsprediction_matrix_mf.tsv- Molecular Function predictions
These files contain raw prediction scores for every protein × GO term combination and can be very large (>50MB).
Note
Use the --skip-matrix flag to skip their generation and only produce the main results file.
Main Results File#
File: results.tsv
The main output file containing functional annotations for all query proteins. This tab-separated file contains the following columns:
Column Descriptions#
protein - Name of the protein from the input FASTA file.
network_type - Type of neural network used for prediction:
gcn(Graph Convolutional Network) - Used when structural information is available from database alignment, providing more confident predictions.cnn(Convolutional Neural Network) - Used when no proteins above similarity cutoff (50% identity by default) are found.
prediction_mode - Ontology category:
mf(Molecular Function),bp(Biological Process),cc(Cellular Component), orec(Enzyme Commission).go_term - Predicted GO term identifier or EC number.
score - DeepFRI confidence score for the prediction. Higher scores indicate greater confidence. See the DeepFRI publication for details.
go_name - Human-readable annotation from the Gene Ontology or EC nomenclature.
aligned - Boolean indicating whether the query was successfully aligned to a database structure (
True/False).target_id - Identifier of the matched database entry (e.g.,
3al6_Dfor PDB chain). Empty if no hit was found.db_name - Name of the database where the match was found (e.g.,
pdb100_230517,afdb_swissprot_v4).query_identity - Sequence identity percentage between query and hit (0.0-1.0 scale). Empty if no hit was found.
query_coverage - Proportion of query sequence covered by the alignment (0.0-1.0 scale).
target_coverage - Proportion of target sequence covered by the alignment (0.0-1.0 scale).
Filtering Recommendations#
Recommended thresholds for filtering predictions:
High confidence: score >= 0.5
Structure-based predictions: network_type == ‘gcn’
Good alignment: query_coverage >= 0.7 and query_identity >= 0.3
Example filtering in Python:
import pandas as pd
# Load results
df = pd.read_csv("results.tsv", sep="\t")
# Filter high-confidence structure-based predictions
filtered = df[
(df["score"] >= 0.5) &
(df["network_type"] == "gcn") &
(df["aligned"] == True)
]
Biological Context#
The protein template is defined as a sequence with at least 50% sequence identity and 90% sequence coverage, as with these parameters we observe mostly the same folds [1]. Majority of proteins will not differ in structure above this threshold [2]. Predicted structures are as useful as experimental ones (Fig. 1a). The bad quality of the structure does decrease the verbosity of predicted GO terms, yet predictions still remain accurate due to the contribution of the sequence module (Fig. 1b, c).
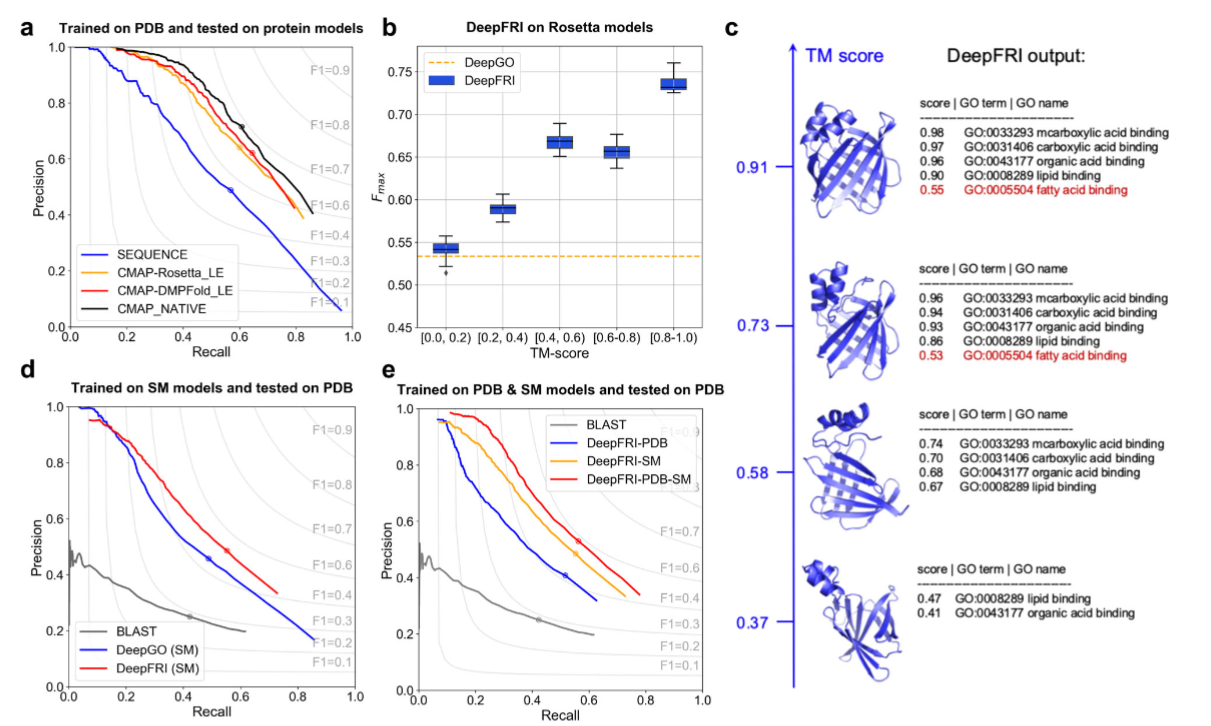
Figure 1. Figure 2 reproduced from original DeepFRI manuscript [3].#
In the follow up work, we demonstrated that DeepFRI predictions are concordant with EggNOG and HUMAnN 2.0 on the samples from cohort of neonatal patients. It should be noted, that structure module was not used in the study, and still was able to generalise to unknown taxa compared to EggNOG. However, EggNOG gave significantly better annotations for class Gammaproteobacteria [4].
Bibliography